Voy a empezar este post explicando un poco q son los ganglios basales asi se va a entender mejor todo lo q se explica mas adelante.
Ganglios basales
Los ganglios basales son acumulaciones de cuerpos de células nerviosas que se hallan cerca de la base del cerebro. Este tejido nervioso gris está interconectado con la corteza cerebral, el tálamo y el tallo cerebral.
En los mamíferos están asociados, fundamentalmente, con los movimientos (que también tienen su origen en la corteza cerebral motora): sus fibras, que no se dirigen directamente a la columna vertebral, enlazan con la corteza cerebral luego de hacer sinapsis con neuronas del talamo. Los ganglios basales se asocian con movimientos voluntarios realizados de forma principalmente inconsciente, esto es, aquellos que involucran al cuerpo entero en tareas rutinarias o cotidianas.
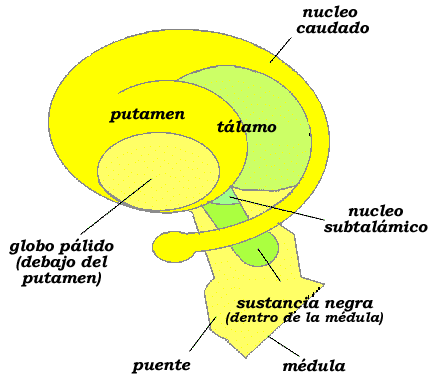
Se sitúan sobre una zona denominada cuerpo estriado: dos cuerpos de sustancia gris separados por un haz de fibras, denominado cápsula interna. Respecto de esta se van situando los ganglios basales: el núcleo caudado, el putamen, el globo pálido, el núcleo subtalámico y la sustancia negra. En el lado interno de la cápsula interna se halla el núcleo caudado (núcleo de la cola) y en su lado externo el putamen (núcleo en forma de cáscara), junto al que se sitúa el globo pálido (una estructura triangular de color gris claro con una fina capa de sustancia blanca en su mitad que, en ocasiones, se une con el putamen para formar el núcleo lentiforme). Situado al lado del globo pálido, pero más hacia el interior, se encuentra el núcleo subtalámico y, por debajo de este, la sustancia negra. Se discute si una delgada tira de sustancia gris situada al lado del putamen, el claustro, del que se desconoce su función, pertenece o no a los ganglios basales: el daño de los ganglios basales implica una falla en la coordinación que supone la aparición de los síntomas característicos de un trastorno motor global; especialmente, los movimientos característicos de enfermedades como el parkinson, el balismo y el corea de Huntington.
Enfermedad de Parkinson
La enfermedad de Parkinson (EP) es una enfermedad neurodegenerativa del sistema nervioso central cuya principal característica es la muerte progresiva de neuronas en una parte del cerebro denominada sustancia negra pars compacta. La consecuencia más importante de esta pérdida neuronal es una marcada disminución en la disponibilidad cerebral de dopamina, principal sustancia sintetizada por estas neuronas, originándose una disfunción en la regulación de las principales estructuras cerebrales implicadas en el control del movimiento.
Causas
No se conoce la causa de esta enfermedad. Recientemente se ha descubierto la existencia de anomalías genéticas en algunas familias en las que la mayoría de sus miembros estaban afectados y en casos familiares de enfermedad de Parkinson de presentación precoz (edad de inicio inferior a 40 años). Sin embargo, la mayoría de los pacientes con enfermedad de Parkinson tienen una presentación esporádica, es decir sin factores genéticos claramente identificados.
Determinados agentes tóxicos son capaces de provocar cuadros clínicos similares a la enfermedad de Parkinson, pero se trata de casos puntuales que no permiten esgrimir una hipótesis tóxica como principal causa de la enfermedad. Existen indicios para pensar que errores del metabolismo energético celular podrían predisponer a determinadas personas a padecer enfermedad de Parkinson, convirtiendo a las neuronas dopaminérgicas en vulnerables a determinados tóxicos ambientales (exógenos) o del propio organismo (endógenos).
En la actualidad se puede resumir que la EP no tiene una causa única, existiendo casos familiares con anomalías genéticas conocidas y otros casos en los que una conjunción de factores genéticos y ambientales serían los responsables de la muerte neuronal.
Sintomas:
La EP se caracteriza por ser un trastorno motor progresivo cuyos principales síntomas son la torpeza generaliza con lentitud en la realización de movimientos, escasez de motilidad espontánea, temblor de reposo y rigidez.
Manifestaciones típicas son la inexpresividad facial («cara de póker»), la escasez de movimientos automáticos como el parpadeo o el braceo al caminar, la inclinación de tronco hacia delante durante la marcha, etc. Conforme aumenta la duración de la enfermedad pueden aparecer otros síntomas, como un deterioro de la marcha con dificultad fundamentalmente al inicio de ésta y en los giros, en los que el paciente se queda como pegado al suelo, fenómeno que se denomina imantación.
También se puede producir una alteración de los reflejos de reequilibración dando lugar a caídas. No es infrecuente la asociación de síntomas no motores como cierto grado de depresión o de disfunción autonómica (urgencia e incontinencia miccional) y en algunos casos, tras muchos de años evolución, la aparición de cierto deterioro cognitivo.
En cualquier caso, debe hacerse énfasis en la enorme variabilidad en la evolución de la enfermedad de Parkinson, de forma que en algunos pacientes el proceso neurodegenerativo progresa muy lentamente mientras que en otros puede ser más rápido.
Edad de aparición:
La edad de máxima presentación es a partir de los 60 años aumentando progresivamente hasta los 80 años. Sin embargo, existen casos de presentación a edades más tempranas, pudiendo ocurrir a cualquier edad a partir de la segunda década. Afecta prácticamente por igual a ambos sexos.
Tratamiento:
El tratamiento fundamental es la reposición de dopamina cerebral mediante la administración farmacológica de su precursor (levodopa). Sin embargo, la administración crónica de este fármaco (sinemet y madopar) varias veces al día se asocia con complicaciones motoras a mediano-largo plazo. Éstas consisten fundamentalmente en fluctuaciones motoras (periodos de mala y buena movilidad alternantes a lo largo del día y en relación a la toma de medicación) y las disquinesias (movimientos involuntarios) durante la fase de buena movilidad. Con el fin de disminuir estas complicaciones se han desarrollado fármacos con acción más prolongada (agonistas dopaminergicos) que consiguen mejorar esta situación de forma transitoria e incompleta, y, recientemente, se han introducido fármacos que prolongan y estabilizan los niveles plasmáticos de levodopa (Comtan, Tasmar).
En los casos en los que las medidas farmacológicas no consiguen controlar los problemas del paciente, el tratamiento quirúrgico mediante lesión o estimulación de estructuras cerebrales malfuncionantes en la EP, tales como el núcleo subtalámico o el globo pálido interno, es otra posibilidad terapéutica con buenos resultados en series amplias de pacientes con años de seguimiento. La utilización de transplantes celulares se encuentra todavía en fase de experimentación en modelos animales.
Pronostico:
La EP es una enfermedad crónica, de larga evolución y curso progresivo. El deterioro motor y las complicaciones en relación a la toma de medicación conllevan un importante grado de incapacidad, aunque como se ha dicho la evolución es variable. En el momento actual el tratamiento quirúrgico mediante estimulación o lesión de núcleo subtalámico o globo palido interno proporciona una gran mejoría en la calidad de vida de estos pacientes.

Enfermedad de Huntington
La enfermedad de Huntington es un trastorno cerebral caracterizado por neurodegeneración progresiva que se manifiesta en disfunciones motoras, cognoscitivas y psiquiátricas. Pueden presentarse problemas en las siguientes tres categorías: control motor (movimiento), cognición (razonamiento) y comportamiento. Ocurren problemas al hablar y al tragar cuando se ven afectados los centros de control motor o cognitivo, lo que causa debilidad muscular o falta de coordinación ( corea ). También se presentan problemas de memoria, secuenciación, capacidad de aprendizaje, razonamiento y resolución de problemas.
Causas
causado por un gen mutante en el brazo corto del cromosoma 4 (gen de huntingtina). Es una repeticion inestable de tripletes: en una persona sana el gen tiene entre 15 y 34 repeticiones mientras q en los pacientes con Huntington tiene entre 42 y 66 repeticiones.
Por culpa de este gen se degeneran las neuronas inhibidoras q se proyectan al segmento externo del globo pálido, por lo tanto estas celulas se tornan anormalmente activas reduciendo las eferencias inhibitorias de los ganglios basales provocando q las neuronas motoras superiores puedan ser activadas por señales inapropiadas.
Edad de aparicion:
Hay dos formas de la enfermedad de Huntington y la más común es la de aparición en la edad adulta. Las personas con esta forma de la enfermedad generalmente presentan síntomas a mediados de la tercera y cuarta década de sus vidas.
Una forma de la enfermedad de Huntington de aparición temprana representa un pequeño número de casos y se inicia en la niñez o en la adolescencia. Los síntomas se pueden parecer a los de la enfermedad de Parkinson con rigidez, movimientos lentos y temblor.
Si uno de sus padres tiene la enfermedad de Huntington, usted tiene un 50% de probabilidad de heredar el gen para dicha enfermedad. Si usted hereda el gen de sus padres, desarrollará la enfermedad en algún momento de su vida y se la puede transmitir a su vez a sus hijos. Si usted no recibe el gen de sus padres, no se lo puede transmitir a sus hijos.
Sintomas
El tipo y la gravedad de los problemas cognitivos y de comunicación varían de persona en persona. A pesar de que muchas cosas pueden parecer similares, no hay dos personas con enfermedad de Huntington que sean exactamente iguales. La siguiente lista resume los problemas que pueden experimentar estas personas durante las distintas etapas de la enfermedad. En muchos casos, la persona experimentará las mismas dificultades durante todo el curso de la enfermedad, y su gravedad variará de etapa en etapa.
Problemas de Comunicación
1. Debilidad muscular, lentitud o falta de coordinación de los labios, la lengua, la garganta y la mandíbula ( disartria)
2. Disminución del control de la velocidad (hablar demasiado rápido o demasiado despacio)
3. Problemas con la calidad de la voz (ronca/áspera, entrecortada, volumen demasiado alto o demasiado bajo)
4. Dificultad en hallar las palabras para expresarse
5."Trabarse" con ciertas palabras o frases, repitiéndolas con frecuencia y en momentos inapropiados (perseveración)
6. Incapacidad de hablar
7. Dificultad para comenzar a decir una palabra u oración, con repetición de sonidos (tartamudez)
8. Dificultad en entender información
9. Dificultad en leer y escribir
10. Disminución de la memoria, inmediata y a corto plazo (por lo general la memoria a largo plazo permanece intacta)
11. Problemas de discernimiento/razonamiento
12. Disminución de la capacidad de aprender nuevas cosas
13. Problemas al Tragar (la principal causa de muerte entre las personas que tienen esta enfermedad es la pulmonía debido a aspiración. Esto puede ocurrir cuando los líquidos o los alimentos entran a las vías respiratorias en vez de al esófago al comer o beber y se acumulan en los pulmones, lo que puede causar pulmonía)
Diagnostico:
La enfermedad de Huntington se diagnostica mediante pruebas genéticas y neurológicas. Se pueden efectuar estas pruebas antes de que la persona exhiba síntoma alguno para determinar si es portadora del gen de Huntington.
¿Cuáles son los tratamientos posibles para la persona con enfermedad de Huntington?
Tratamiento:
Por lo general, el médico receta al paciente medicinas para aliviar los síntomas. Hasta la fecha no hay cura para la enfermedad ni forma alguna de detener el progreso de la misma.
Por ultimo... cualquier duda q tengan (y este dentro de mis posibilidades responderla) no duden en preguntar.
Saludos!!!
Fuentes:
http://www.geosalud.com/adultos_mayores/parkinson.htm
http://www.asha.org/public/speech/disorders/huntington.htm
http://es.wikipedia.org/wiki/Ganglios_basales
Neurociencia 3ª edicion, Dale Purves
Ganglios basales
Los ganglios basales son acumulaciones de cuerpos de células nerviosas que se hallan cerca de la base del cerebro. Este tejido nervioso gris está interconectado con la corteza cerebral, el tálamo y el tallo cerebral.
En los mamíferos están asociados, fundamentalmente, con los movimientos (que también tienen su origen en la corteza cerebral motora): sus fibras, que no se dirigen directamente a la columna vertebral, enlazan con la corteza cerebral luego de hacer sinapsis con neuronas del talamo. Los ganglios basales se asocian con movimientos voluntarios realizados de forma principalmente inconsciente, esto es, aquellos que involucran al cuerpo entero en tareas rutinarias o cotidianas.
Se sitúan sobre una zona denominada cuerpo estriado: dos cuerpos de sustancia gris separados por un haz de fibras, denominado cápsula interna. Respecto de esta se van situando los ganglios basales: el núcleo caudado, el putamen, el globo pálido, el núcleo subtalámico y la sustancia negra. En el lado interno de la cápsula interna se halla el núcleo caudado (núcleo de la cola) y en su lado externo el putamen (núcleo en forma de cáscara), junto al que se sitúa el globo pálido (una estructura triangular de color gris claro con una fina capa de sustancia blanca en su mitad que, en ocasiones, se une con el putamen para formar el núcleo lentiforme). Situado al lado del globo pálido, pero más hacia el interior, se encuentra el núcleo subtalámico y, por debajo de este, la sustancia negra. Se discute si una delgada tira de sustancia gris situada al lado del putamen, el claustro, del que se desconoce su función, pertenece o no a los ganglios basales: el daño de los ganglios basales implica una falla en la coordinación que supone la aparición de los síntomas característicos de un trastorno motor global; especialmente, los movimientos característicos de enfermedades como el parkinson, el balismo y el corea de Huntington.
Enfermedad de Parkinson
La enfermedad de Parkinson (EP) es una enfermedad neurodegenerativa del sistema nervioso central cuya principal característica es la muerte progresiva de neuronas en una parte del cerebro denominada sustancia negra pars compacta. La consecuencia más importante de esta pérdida neuronal es una marcada disminución en la disponibilidad cerebral de dopamina, principal sustancia sintetizada por estas neuronas, originándose una disfunción en la regulación de las principales estructuras cerebrales implicadas en el control del movimiento.
Causas
No se conoce la causa de esta enfermedad. Recientemente se ha descubierto la existencia de anomalías genéticas en algunas familias en las que la mayoría de sus miembros estaban afectados y en casos familiares de enfermedad de Parkinson de presentación precoz (edad de inicio inferior a 40 años). Sin embargo, la mayoría de los pacientes con enfermedad de Parkinson tienen una presentación esporádica, es decir sin factores genéticos claramente identificados.
Determinados agentes tóxicos son capaces de provocar cuadros clínicos similares a la enfermedad de Parkinson, pero se trata de casos puntuales que no permiten esgrimir una hipótesis tóxica como principal causa de la enfermedad. Existen indicios para pensar que errores del metabolismo energético celular podrían predisponer a determinadas personas a padecer enfermedad de Parkinson, convirtiendo a las neuronas dopaminérgicas en vulnerables a determinados tóxicos ambientales (exógenos) o del propio organismo (endógenos).
En la actualidad se puede resumir que la EP no tiene una causa única, existiendo casos familiares con anomalías genéticas conocidas y otros casos en los que una conjunción de factores genéticos y ambientales serían los responsables de la muerte neuronal.
Sintomas:
La EP se caracteriza por ser un trastorno motor progresivo cuyos principales síntomas son la torpeza generaliza con lentitud en la realización de movimientos, escasez de motilidad espontánea, temblor de reposo y rigidez.
Manifestaciones típicas son la inexpresividad facial («cara de póker»), la escasez de movimientos automáticos como el parpadeo o el braceo al caminar, la inclinación de tronco hacia delante durante la marcha, etc. Conforme aumenta la duración de la enfermedad pueden aparecer otros síntomas, como un deterioro de la marcha con dificultad fundamentalmente al inicio de ésta y en los giros, en los que el paciente se queda como pegado al suelo, fenómeno que se denomina imantación.
También se puede producir una alteración de los reflejos de reequilibración dando lugar a caídas. No es infrecuente la asociación de síntomas no motores como cierto grado de depresión o de disfunción autonómica (urgencia e incontinencia miccional) y en algunos casos, tras muchos de años evolución, la aparición de cierto deterioro cognitivo.
En cualquier caso, debe hacerse énfasis en la enorme variabilidad en la evolución de la enfermedad de Parkinson, de forma que en algunos pacientes el proceso neurodegenerativo progresa muy lentamente mientras que en otros puede ser más rápido.
Edad de aparición:
La edad de máxima presentación es a partir de los 60 años aumentando progresivamente hasta los 80 años. Sin embargo, existen casos de presentación a edades más tempranas, pudiendo ocurrir a cualquier edad a partir de la segunda década. Afecta prácticamente por igual a ambos sexos.
Tratamiento:
El tratamiento fundamental es la reposición de dopamina cerebral mediante la administración farmacológica de su precursor (levodopa). Sin embargo, la administración crónica de este fármaco (sinemet y madopar) varias veces al día se asocia con complicaciones motoras a mediano-largo plazo. Éstas consisten fundamentalmente en fluctuaciones motoras (periodos de mala y buena movilidad alternantes a lo largo del día y en relación a la toma de medicación) y las disquinesias (movimientos involuntarios) durante la fase de buena movilidad. Con el fin de disminuir estas complicaciones se han desarrollado fármacos con acción más prolongada (agonistas dopaminergicos) que consiguen mejorar esta situación de forma transitoria e incompleta, y, recientemente, se han introducido fármacos que prolongan y estabilizan los niveles plasmáticos de levodopa (Comtan, Tasmar).
En los casos en los que las medidas farmacológicas no consiguen controlar los problemas del paciente, el tratamiento quirúrgico mediante lesión o estimulación de estructuras cerebrales malfuncionantes en la EP, tales como el núcleo subtalámico o el globo pálido interno, es otra posibilidad terapéutica con buenos resultados en series amplias de pacientes con años de seguimiento. La utilización de transplantes celulares se encuentra todavía en fase de experimentación en modelos animales.
Pronostico:
La EP es una enfermedad crónica, de larga evolución y curso progresivo. El deterioro motor y las complicaciones en relación a la toma de medicación conllevan un importante grado de incapacidad, aunque como se ha dicho la evolución es variable. En el momento actual el tratamiento quirúrgico mediante estimulación o lesión de núcleo subtalámico o globo palido interno proporciona una gran mejoría en la calidad de vida de estos pacientes.
Enfermedad de Huntington
La enfermedad de Huntington es un trastorno cerebral caracterizado por neurodegeneración progresiva que se manifiesta en disfunciones motoras, cognoscitivas y psiquiátricas. Pueden presentarse problemas en las siguientes tres categorías: control motor (movimiento), cognición (razonamiento) y comportamiento. Ocurren problemas al hablar y al tragar cuando se ven afectados los centros de control motor o cognitivo, lo que causa debilidad muscular o falta de coordinación ( corea ). También se presentan problemas de memoria, secuenciación, capacidad de aprendizaje, razonamiento y resolución de problemas.
Causas
causado por un gen mutante en el brazo corto del cromosoma 4 (gen de huntingtina). Es una repeticion inestable de tripletes: en una persona sana el gen tiene entre 15 y 34 repeticiones mientras q en los pacientes con Huntington tiene entre 42 y 66 repeticiones.
Por culpa de este gen se degeneran las neuronas inhibidoras q se proyectan al segmento externo del globo pálido, por lo tanto estas celulas se tornan anormalmente activas reduciendo las eferencias inhibitorias de los ganglios basales provocando q las neuronas motoras superiores puedan ser activadas por señales inapropiadas.
Edad de aparicion:
Hay dos formas de la enfermedad de Huntington y la más común es la de aparición en la edad adulta. Las personas con esta forma de la enfermedad generalmente presentan síntomas a mediados de la tercera y cuarta década de sus vidas.
Una forma de la enfermedad de Huntington de aparición temprana representa un pequeño número de casos y se inicia en la niñez o en la adolescencia. Los síntomas se pueden parecer a los de la enfermedad de Parkinson con rigidez, movimientos lentos y temblor.
Si uno de sus padres tiene la enfermedad de Huntington, usted tiene un 50% de probabilidad de heredar el gen para dicha enfermedad. Si usted hereda el gen de sus padres, desarrollará la enfermedad en algún momento de su vida y se la puede transmitir a su vez a sus hijos. Si usted no recibe el gen de sus padres, no se lo puede transmitir a sus hijos.
Sintomas
El tipo y la gravedad de los problemas cognitivos y de comunicación varían de persona en persona. A pesar de que muchas cosas pueden parecer similares, no hay dos personas con enfermedad de Huntington que sean exactamente iguales. La siguiente lista resume los problemas que pueden experimentar estas personas durante las distintas etapas de la enfermedad. En muchos casos, la persona experimentará las mismas dificultades durante todo el curso de la enfermedad, y su gravedad variará de etapa en etapa.
Problemas de Comunicación
1. Debilidad muscular, lentitud o falta de coordinación de los labios, la lengua, la garganta y la mandíbula ( disartria)
2. Disminución del control de la velocidad (hablar demasiado rápido o demasiado despacio)
3. Problemas con la calidad de la voz (ronca/áspera, entrecortada, volumen demasiado alto o demasiado bajo)
4. Dificultad en hallar las palabras para expresarse
5."Trabarse" con ciertas palabras o frases, repitiéndolas con frecuencia y en momentos inapropiados (perseveración)
6. Incapacidad de hablar
7. Dificultad para comenzar a decir una palabra u oración, con repetición de sonidos (tartamudez)
8. Dificultad en entender información
9. Dificultad en leer y escribir
10. Disminución de la memoria, inmediata y a corto plazo (por lo general la memoria a largo plazo permanece intacta)
11. Problemas de discernimiento/razonamiento
12. Disminución de la capacidad de aprender nuevas cosas
13. Problemas al Tragar (la principal causa de muerte entre las personas que tienen esta enfermedad es la pulmonía debido a aspiración. Esto puede ocurrir cuando los líquidos o los alimentos entran a las vías respiratorias en vez de al esófago al comer o beber y se acumulan en los pulmones, lo que puede causar pulmonía)
Diagnostico:
La enfermedad de Huntington se diagnostica mediante pruebas genéticas y neurológicas. Se pueden efectuar estas pruebas antes de que la persona exhiba síntoma alguno para determinar si es portadora del gen de Huntington.
¿Cuáles son los tratamientos posibles para la persona con enfermedad de Huntington?
Tratamiento:
Por lo general, el médico receta al paciente medicinas para aliviar los síntomas. Hasta la fecha no hay cura para la enfermedad ni forma alguna de detener el progreso de la misma.
Por ultimo... cualquier duda q tengan (y este dentro de mis posibilidades responderla) no duden en preguntar.
Saludos!!!
Fuentes:
http://www.geosalud.com/adultos_mayores/parkinson.htm
http://www.asha.org/public/speech/disorders/huntington.htm
http://es.wikipedia.org/wiki/Ganglios_basales
Neurociencia 3ª edicion, Dale Purves