Bueno compañeros taringueros recientemente vi un video de una banda conocida como Sigur Ros (no pregunten mucho sobre ella, apenas la conozco) que tenia un video muy interesante en el que actuaban chicos con sindrome de down. Lo que me llevo a preguntarme si habia informacion al respecto en T! pero al usar el buscador no halle nada asi que aqui va, adicionalmente va el video de Sigur Ros y algunas frases sobre musica que estan en el video que de paso se las traduje.
La idea de este post es que aprendamos a respetar a los demás sea que tengan sindrome de down, o pertenezcan a una minoría...
Síndrome de Down
El síndrome de Down (SD) es un trastorno genético causado por la presencia de tres cromosomas 21, en vez de los dos habituales (trisomía del par 21), caracterizado por la presencia de un grado variable de retraso mental y unos rasgos físicos peculiares que le dan un aspecto reconocible. Es la causa más frecuente de discapacidad psíquica congénita y debe su nombre a John Langdon Haydon Down que fue el primero en describir esta condición en 1866, aunque nunca llegó a descubrir las causas que la producían. En julio de 1958 un joven investigador llamado Jérôme Lejeune descubrió que el síndrome es una alteración en el mencionado par de cromosomas.
No se conocen con exactitud las causas que provocan el exceso cromosómico, aunque se relaciona estadísticamente con una edad materna superior a los 35 años. Las personas con Síndrome de Down tienen una probabilidad algo superior a la de la población general de padecer algunas patologías, especialmente de corazón, sistema digestivo y sistema endocrino, debido al exceso de proteínas sintetizadas por el cromosoma de más. Los avances actuales en el descifrado del genoma humano están desvelando algunos de los procesos bioquímicos subyacentes al retraso mental, pero en la actualidad no existe ningún tratamiento farmacológico que haya demostrado mejorar las capacidades intelectuales de estas personas. Las terapias de estimulación precoz y el cambio en la mentalidad de la sociedad, por el contrario, sí están suponiendo un cambio cualitativo positivo en sus expectativas vitales.
Genética
Las células del ser humano poseen cada una en su núcleo 23 pares de cromosomas. Cada progenitor aporta a su descendencia la mitad de la información genética, en forma de un cromosoma de cada par. 22 de esos pares se denominan autosomas y el último corresponde a los cromosomas sexuales (X o Y).
Tradicionalmente los pares de cromosomas se describen y nombran en función de su tamaño, del par 1 al 22 (de mayor a menor), más el par de cromosomas sexuales antes mencionado. El cromosoma 21 es el más pequeño, en realidad, por lo que debería ocupar el lugar 22, pero un error en la convención de Denver del año 1960, que asignó el síndrome de Down al par 21 ha perdurado hasta nuestros días, manteniéndose por razones prácticas esta nomenclatura.
El cromosoma 21 contiene aproximadamente el 1% de la información genética de un individuo en algo más de 400 genes, aunque hoy en día sólo se conoce con precisión la función de unos pocos.
En la imagen podemos observar un Cariotipo (conjunto de cromosomas de un individuo) mostrando una trisomía libre del par.
Trisomía libre
El síndrome de Down se produce por la aparición de un cromosoma más en el par 21 original (tres cromosomas: “trisomía” del par 21) en las células del organismo. La nomenclatura científica para ese exceso cromosómico es 47, XX,+21 o 47, XY,+21; según se trate de una mujer o de un varón, respectivamente. La mayor parte de las personas con este síndrome (95%), deben el exceso cromosómico a un error durante la primera división meiótica (aquella por la que los gametos, óvulos o espermatozoides, pierden la mitad de sus cromosomas) llamándose a esta variante, “trisomía libre” o regular. El error se debe en este caso a una disyunción incompleta del material genético de uno de los progenitores. (En la formación habitual de los gametos el par de cromosomas se separa, de modo que cada progenitor sólo transmite la información de uno de los cromosomas de cada par. Cuando no se produce la disyunción se transmiten ambos cromosomas).
No se conocen con exactitud las causas que originan la disyunción errónea. Como en otros procesos similares se han propuesto hipótesis multifactoriales (exposición ambiental, envejecimiento celular…) sin que se haya conseguido establecer ninguna relación directa entre ningún agente causante y la aparición de la trisomía. El único factor que presenta una asociación estadística estable con el síndrome es la edad materna, lo que parece apoyar las teorías que hacen hincapié en el deterioro del material genético con el paso del tiempo.
Translocación del brazo corto del cromosoma 21 en uno de los dos cromosomas del par 14.
Translocación del brazo corto del cromosoma 21 en uno de los dos cromosomas del par 14.
En aproximadamente un 15% de los casos el cromosoma extra es transmitido por el espermatozoide y en el 85% restante por el óvulo.
Translocación
Después de la trisomía libre, la causa más frecuente de aparición del exceso de material genético es la translocación. En esta variante el cromosoma 21 extra (o un fragmento del mismo) se encuentra “pegado” a otro cromosoma (frecuentemente a uno de los dos cromosomas del par 14), por lo cual el recuento genético arroja una cifra de 46 cromosomas en cada célula. En este caso no existe un problema con la disyunción cromosómica, pero uno de ellos porta un fragmento “extra” con los genes del cromosoma “translocado”. A efectos de información genética sigue tratándose de una trisomía 21 ya que se duplica la dotación genética de ese cromosoma.
La frecuencia de esta variante es aproximadamente de un 3% de todos los SD y su importancia estriba en la necesidad de hacer un estudio genético a los progenitores para comprobar si uno de ellos era portador sin saberlo de la translocación, o si ésta se produjo por primera vez en el embrión. (Existen portadores “sanos” de translocaciones, en los que se recuentan 45 cromosomas, estando uno de ellos translocado, o pegado, a otro).
Mosaicismo
La forma menos frecuente de trisomía 21 es la denominada “mosaico” (en torno al 2% de los casos). Esta mutación se produce tras la concepción, por lo que la trisomía no está presente en todas las células del individuo con SD, sino sólo en aquellas cuya estirpe procede de la primera célula mutada. El porcentaje de células afectadas puede abarcar desde unas pocas a casi todas, según el momento en que se haya producido la segregación anómala de los cromosomas homólogos.
Expresión del exceso de material genético (o sea lo que provoca este síndrome biológicamente)
La expresión bioquímica del síndrome consiste en el aumento de diferentes enzimas. Una de las más conocidas e importantes es la superóxidodimutasa (codificada por el gen SOD-1), que cataliza el paso del anión superóxido hacia peróxido de hidrógeno. En condiciones normales esto contribuye al sistema de defensa antioxidante del organismo, pero su exceso determina la acumulación de H2O2, lo que puede provocar peroxidación de lípidos y proteínas y dañar el ADN. Otros genes implicados en la aparición de trastornos asociados al SD son:
* COL6A1: Su expresión incrementada se relaciona con defectos cardiacos
* ETS2: Su expresión incrementada puede ser causa de alteraciones músculo esqueléticas
* CAF1A: La presencia incrementada de este gen puede interferir en la síntesis de ADN
* Cystathione Beta Synthase (CBS): Su exceso puede causar alteraciones metabólicas y de los procesos de reparación del ADN
* DYRK: En el exceso de proteínas codificadas por este gen parece estar el origen del retraso mental
* CRYA1: Su sobreexpresión puede originar cataratas (opacidad precoz del cristalino)
* GART: La expresión aumentada de este gen puede alterar los procesos de síntesis y reparación del ADN
* IFNAR : Es un gen relacionado con la síntesis de Interferón, por lo que su exceso puede provocar alteraciones en el sistema inmunitario.
Cuadro Clínico
El SD es la causa más frecuente de discapacidad psíquica congénita. Representa el 25% de todos los casos de retraso mental. Se trata de un síndrome genético más que de una enfermedad según el modelo clásico, y aunque sí se asocia con frecuencia a algunas patologías, la expresión fenotípica final es muy variada de unas personas a otras. Como rasgos comunes se pueden reseñar su fisiognomía peculiar, una hipotonía muscular generalizada, un grado variable de retraso mental y retardo en el crecimiento.
En cuanto al fenotipo han sido descritos más de 100 rasgos peculiares asociados al SD, pudiendo presentarse en un individuo un número muy variable de ellos. De hecho ninguno se considera constante o patognomónico aunque la evaluación conjunta de los que aparecen suele ser suficiente para el diagnóstico.
Algunos de los rasgos más importantes son un perfil facial y occipital planos, braquiocefalia (predominio del diámetro transversal de la cabeza), hendiduras palpebrales oblicuas, diastasis de rectos (laxitud de la musculatura abdominal), raíz nasal deprimida, pliegues epicánticos (pliegue de piel en el canto interno de los ojos), cuello corto y ancho con exceso de pliegue epidérmico nucal, microdoncia, paladar ojival, clinodactilia del quinto dedo de las manos (crecimiento recurvado hacia el dedo anular), pliegue palmar único, y separación entre el primer y segundo dedo del pie. Las patologías que se asocian con más frecuencia son las cardiopatías congénitas y enfermedades del tracto digestivo (celiaquía, atresia/estenosis esofágica o duodenal, colitis ulcerosa...). Los únicos rasgos presentes en todos los casos son la atonía muscular generalizada (falta de un tono muscular adecuado, lo que dificulta el aprendizaje motriz) y el retraso mental aunque en grados muy variables.
Presentan, además, un riesgo superior al de la población general, para el desarrollo de patologías como leucemia (leucemia mieloide aguda), diabetes, hipotiroidismo, miopía, o luxación atloaxoidea (inestabilidad de la articulación entre las dos primeras vértebras, atlas y axis, secundaria a la hipotonía muscular y a la laxitud ligamentosa). Todo esto determina una media de esperanza de vida entre los 50 y los 60 años, aunque este promedio se obtiene de una amplia horquilla interindividual (las malformaciones cardíacas graves o la leucemia, cuando aparecen, son causa de muerte prematura). El grado de discapacidad intelectual también es muy variable, aunque se admite como hallazgo constante un retraso mental ligero o moderado. No existe relación alguna entre los rasgos externos y el desarrollo intelectual de la persona con SD.
Epidemiologia
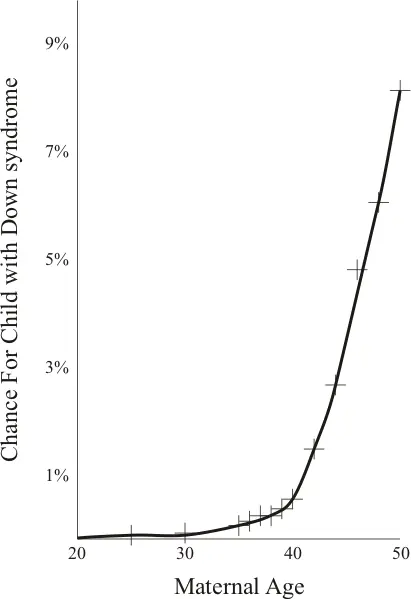
La incidencia global del síndrome de Down se aproxima a uno de cada 700 nacimientos (15/10.000), pero el riesgo varía con la edad de la madre. La incidencia en madres de 25 años es de 1 por cada 2000 nacidos vivos, mientras que en madres de 35 años es de 1 por cada 200 nacimientos y de 1 por cada 40 en las mujeres mayores de 40 años. Por este motivo se recomiendan técnicas de diagnóstico prenatal a todas las mujeres a partir de los 35 años. El ECEMC (Estudio Colaborativo Español de Malformaciones Congénitas) informaba en el año 2004 de una prevalencia neonatal de 7,11 cada 10.000 recién nacidos, con tendencia a disminuir de manera estadísticamente significativa. Esta tendencia, junto con el aumento relativo de casos en mujeres por debajo de 35 años, se atribuye al aumento de interrupciones voluntarias del embarazo tras el diagnóstico prenatal en mujeres por encima de esa edad. Parece existir una relación estadística (sin que se conozcan los mecanismos exactos) entre algunas enfermedades maternas como hepatitis, Mycoplasma hominis tipo 1, Herpes simple tipo II y diabetes y un aumento en la incidencia de aparición de SD; no obstante esa relación estadística no es tan intensa como en el caso de la edad materna. Algún autor también ha relacionado la baja frecuencia coital, así como el uso de anovulatorios y espermicidas con la aparición del síndrome.
Haber tenido un hijo con SD también aumenta la probabilidad de tener otro: el riesgo aumenta de modo general a 1 de cada 100 recién nacidos vivos. Los antecedentes familiares igualmente incrementan ese riesgo. Los varones con síndrome de Down se consideran estériles, pero las mujeres conservan con frecuencia su capacidad reproductiva. En su caso también se incrementa la probabilidad de engendrar hijos con SD.
DIAGNOSTICO
A partir de 1979 se dispone en los laboratorios de una prueba en sangre que permite establecer una sospecha diagnóstica para varios defectos congénitos (espina bífida y otros defectos del tubo neural). Esta prueba es la determinación de los valores de AFP (Alfa-fetoproteína), que se encuentran aumentados en los embriones que presentan estos trastornos del desarrollo. Varios años después se establece una relación estadística entre valores bajos de esta proteína y la aparición de trastornos cromosómicos, en especial del SD. En años posteriores se descubrieron algunas asociaciones similares con otras sustancias en sangre materna. Hoy día es común la determinación de AFP, estriol y hCG (Gonadotropina coriónica humana) para determinar el riesgo de aparición del SD. A esto se le llama “triple prueba”. Algunos laboratorios incluyen la determinación de inhibina (cuádruple prueba). Los valores de estas sustancias en sangre, así como datos acerca de la edad materna y los antecedentes personales y familiares permiten calcular un riesgo de aparición de SD, pero no suponen un diagnóstico de certeza. Determinadas mediciones que se realizan durante las ecografías (longitud del fémur, grosor del pliegue nucal, y otras) también aportan información para el cálculo de ese riesgo, pero tampoco permiten establecer el diagnóstico definitivo.
Para detectar la anormalidad cromosómica durante el periodo prenatal de forma inequívoca se emplean técnicas de conteo cromosómico, por lo que es necesario disponer de alguna célula fetal. El acceso al material celular embrionario puede suponer un cierto riesgo, tanto para la madre como para el feto, por lo que su indicación se circunscribe a aquellos embarazos en los que se haya detectado un riesgo de aparición de la trisomía superior al de la población general (triple prueba positiva, edad materna superior a 35 años o paterna superior a 50, antecedentes familiares o personales de SD, o progenitores portadores de una traslocación equilibrada u otras alteraciones cromosómicas).
La técnica más frecuentemente utilizada para la obtención de material genético fetal es la Amniocentesis. Esta técnica se empezó a generalizar en la década de los 60, y consiste en la punción ecoguiada de la cavidad amniótica por vía abdominal. Se consigue así una muestra de líquido amniótico, de donde es posible obtener células fetales para su estudio. Debe realizarse preferentemente entre las semanas 14 a 17 del embarazo. Es una técnica relativamente inocua y poco molesta pero comporta un riesgo del 1-2% de aborto, lesión fetal, o infección materna.
A mediados de los 80 se comenzó a usar otra técnica, denominada Biopsia de vellosidades coriónicas: se obtiene un fragmento de material placentario por vía vaginal o a través del abdomen, normalmente entre las semanas 8 y 11 del embarazo. Esta técnica se puede realizar antes de que exista la suficiente cantidad de líquido amniótico necesaria para que se pueda llevar a cabo la amniocentesis, y el estudio cromosómico es más rápido pues no se necesita el cultivo celular para obtener una muestra suficientemente grande. Presenta un riesgo para la madre y el feto similar al de la amniocentesis.
TRATAMIENTO
La mejoría en los tratamientos de las enfermedades asociadas al SD ha aumentado la esperanza de vida de estas personas, desde los 14 años hace unas décadas, hasta casi la normalidad (60 años, en países desarrollados) en la actualidad. A lo largo de los últimos 150 años se han postulado diferentes tratamientos empíricos (hormona tiroidea, hormona del crecimiento, ácido glutámico, dimetilsulfóxido, complejos vitamínicos y minerales, 5-hidroxitriptófano o piracetam) sin que ninguno haya demostrado en estudios longitudinales a doble ciego que su administración provoque ningún efecto positivo significativo en el desarrollo motor, social, intelectual o de expresión verbal de las personas con SD. No existe hasta la fecha ningún tratamiento farmacológico eficaz para el SD, aunque los estudios puestos en marcha con la secuenciación del genoma humano permiten augurar una posible vía de actuación (enzimática o genética), eso sí, en un futuro todavía algo lejano.
Los únicos tratamientos que han demostrado una influencia significativa en el desarrollo de los niños con SD son los programas de Atención Temprana, orientados a la estimulación precoz del sistema nervioso central durante los seis primeros años de vida. Especialmente durante los dos primeros años el SNC presenta un grado de plasticidad muy alto lo que resulta útil para potenciar mecanismos de aprendizaje y de comportamiento adaptativo. Los individuos con grandes dificultades para el aprendizaje a menudo han sido internados en instituciones, pero se ha comprobado que deben vivir en su domicilio, donde desarrollan de forma más completa todo su potencial. La adaptación curricular permite en muchos casos una integración normalizada en colegios habituales, aunque deben tenerse en cuenta sus necesidades educativas especiales. La edad mental que pueden alcanzar está todavía por descubrir, y depende directamente del ambiente educativo y social en el que se desarrollan. Cuando éste es demasiado protector, los chicos y chicas tienden (al igual que nos ocurriría a todos) a dejarse llevar, descubriendo escasamente sus potencialidades. Los contextos estimulantes ayudan a que se generen conductas de superación que impulsan el desarrollo de la inteligencia. Como consecuencia, es imposible determinar los trabajos y desempeños que pueden conseguir durante la vida adulta. Potenciar sus iniciativas y romper con los planteamientos estáticos que históricamente les han perseguido son compromisos sociales ineludibles que las sociedades actuales deben atender.
Atencion Temprana
Todos los niños precisan de estímulos para el correcto desarrollo de sus capacidades motrices, cognitivas, emocionales y adaptativas. Los niños con SD no son una excepción, aunque sus procesos de percepción y adquisición de conocimientos son algo diferentes a los del resto de la población: Las capacidades visuales de los niños con SD son, por ejemplo, superiores a las auditivas, y su capacidad comprensiva es superior a la de expresión, por lo que su lenguaje es escaso y aparece con cierto retraso, aunque compensan sus deficiencias verbales con aptitudes más desarrolladas en lenguaje no verbal, como el contacto visual, la sonrisa social o el empleo de señas para hacerse entender. La atonía muscular determina también diferencias en el desarrollo de la habilidad de caminar, o en la motricidad fina. Todos esos aspectos deben ser contemplados en programas específicos de atención temprana (durante los primeros seis años de vida) para estimular al máximo los mecanismos adaptativos y de aprendizaje más apropiados. Intentar enseñar a leer a un niño con SD utilizando métodos convencionales, por ejemplo, puede convertirse en una tarea muy difícil, si no se tiene en cuenta su superior capacidad visual. Hoy día existen métodos gráficos (a partir de tarjetas, o fichas, que asocian imagen y palabra) que están consiguiendo resultados muy superiores al clásico encadenado de letras en estos niños. Además el objetivo de estos programas no es tan sólo la adquisición de habilidades, sino que estas se alcancen mucho antes, permitiendo continuar con programas educativos que integren al máximo a la persona con SD en entornos normalizados
[b○3Pronosticos y expectativas de futuro[/b]
Se desconocen todavía los mecanismos que provocan el retraso mental en las personas con SD, aunque la secuenciación del genoma humano y diversos estudios llevados a cabo en sujetos con translocaciones parciales están empezando a servir para descubrir los genes responsables del cuadro. Estos mapas fenotípicos también se han comparado con algunos casos de monosomía 21 (cuadro de ausencia de uno de los dos cromosomas del par 21, la situación contraria al SD) obteniéndose así mapas de rasgos asociados al exceso o defecto de dosis cromosómica. [36] En las próximas décadas todo este conocimiento sobre el funcionamiento y expresión de los genes permitirá, con seguridad, establecer nuevas estrategias terapéuticas capaces de revertir los trastornos cognitivos asociados al síndrome de Down, y muchos de sus problemas asociados.
En 1981 se diseñó el primer Programa de Salud específico para personas con SD, pero el más ampliamente aceptado y difundido en la comunidad científica es el diseñado por el Down Syndrome Medical Interest group (DSMIG) . En estos programas de salud se contemplan las actuaciones preventivas mínimas para un adecuado diagnóstico precoz y seguimiento de las enfermedades o complicaciones que se pueden presentar, mejorando significativamente el pronóstico de estas personas. Por otra parte los programas, cada vez más extendidos, de estimulación precoz, y el cambio progresivo de mentalidad que la sociedad está experimentando con respecto a la discapacidad intelectual son los principales motivos de la gran transformación que se está viviendo en torno a las personas con SD. Hace apenas unas décadas estas personas eran apartadas de la sociedad en instituciones, o escondidas por sus progenitores, en base a un falso complejo de culpa. A pesar del enorme esfuerzo que aún queda pendiente hoy podemos comprobar cómo un entorno basado en la aceptación, en la adaptación de los métodos de aprendizaje y en la virtud de la diversidad está dotando a las personas con SD de la autonomía suficiente como para trabajar, vivir en pareja o desarrollar habilidades artísticas impensables hace muy poco tiempo
Ufff ahora si el video de Sigur Ros
SIGUR ROS - Svefn-g-englar
link: http://www.videos-star.com/watch.php?video=zQ5Grncdjlc
Frases al principio del video
"Sin música la vida seria una equivocación" (Friedrich Nietzche)
"La música fue inventada para confirmar la soledad humana" (Lawrence Dureeus)
"La música es la forma rápida y abreviada de las emociones" [Leo Toisley)
"La música expresa aquella que no puede ser dicho en lo que es imposible de callar" (Victor Hugo)
"La música es una ley moral, le da alma al universo, alas a la mente, vuelo a la imaginación, encanto y tranquilidad a la vida y a todo" (Platón)
"Un pintor pinta imágenes en lienzos. Pero los músicos pintan su música en el silencio" (Nombre Ilegible)
"Si miras lo suficientemente profundo, verás musicalmente. El corazón de la Naturaleza siendo música en todos lados" (Thomas Cartile)
"La música nombra lo innombrable y comunica lo imposible de conocer" (Leonard Bernstein)
"...En el final veo a la música como la gracia de salvación para toda la humanidad" (Henry Millers)
"La música es bien nombrada como el habla de los ángeles, de hecho nada entre los (esta palabra se refiere a un acto no verbal en el que se transmite un mensaje, como un lenguaje no hablado) permitidos al hombre se siente tan divino. Nos lleva cerca del infinito" (Thomas Carlyle)
Ahora si la fuente... es wikipedia pero citare las fuentes que usaron los users en wikipedia, he revisado y son confiables entre a los links y revise si estan muertos y arregle algunos que estaban mal, todas las fuentes 100% confiables y comprobadas por moi
DADO LO EXTENSO ME HE TOMADO LA LIBERTAD DE RESALTAR ALGUNOS ASPECTOS QUE DEBERIAN SER LEIDOS.
* Siegfried M. Pueschel (2002) Síndrome de Down: Hacia un futuro mejor, Ed. Masson ISBN 1-55766-452-8.
* Down, J.H.L. (1886). Observations on an ethnic classification of idiots. London Hospital. Clinical Lectures and Reports, 3: 259-262.
* Josep M. Corretger et al (2005). Síndrome de Down: Aspectos médicos actuales. Ed. Masson, para la Fundación Catalana del Síndrome de Down. ISBN 84-458-1504-0.
* Azucena Martínez Acebal, Joaquín Fernández Toral (1999). Síndrome de Down: Aspectos sociológicos, Médicos y Legales. ISBN 84-86889-65-0.
* Pilar Arranz Martínez (2002). Niños y jóvenes con Síndrome de Down. Egido Editorial. ISBN 84-95879-09-3.
* Candel, I. Programa de Atención temprana. Intervención en niños con síndrome de Down y otros problemas del desarrollo. Ed. CEPE, Madrid, 1999.
http://www.ctdownsyndrome.org/index.php?option=com_content&task=view&id=73&Itemid=118
La idea de este post es que aprendamos a respetar a los demás sea que tengan sindrome de down, o pertenezcan a una minoría...
Síndrome de Down
El síndrome de Down (SD) es un trastorno genético causado por la presencia de tres cromosomas 21, en vez de los dos habituales (trisomía del par 21), caracterizado por la presencia de un grado variable de retraso mental y unos rasgos físicos peculiares que le dan un aspecto reconocible. Es la causa más frecuente de discapacidad psíquica congénita y debe su nombre a John Langdon Haydon Down que fue el primero en describir esta condición en 1866, aunque nunca llegó a descubrir las causas que la producían. En julio de 1958 un joven investigador llamado Jérôme Lejeune descubrió que el síndrome es una alteración en el mencionado par de cromosomas.
No se conocen con exactitud las causas que provocan el exceso cromosómico, aunque se relaciona estadísticamente con una edad materna superior a los 35 años. Las personas con Síndrome de Down tienen una probabilidad algo superior a la de la población general de padecer algunas patologías, especialmente de corazón, sistema digestivo y sistema endocrino, debido al exceso de proteínas sintetizadas por el cromosoma de más. Los avances actuales en el descifrado del genoma humano están desvelando algunos de los procesos bioquímicos subyacentes al retraso mental, pero en la actualidad no existe ningún tratamiento farmacológico que haya demostrado mejorar las capacidades intelectuales de estas personas. Las terapias de estimulación precoz y el cambio en la mentalidad de la sociedad, por el contrario, sí están suponiendo un cambio cualitativo positivo en sus expectativas vitales.
Genética
Las células del ser humano poseen cada una en su núcleo 23 pares de cromosomas. Cada progenitor aporta a su descendencia la mitad de la información genética, en forma de un cromosoma de cada par. 22 de esos pares se denominan autosomas y el último corresponde a los cromosomas sexuales (X o Y).
Tradicionalmente los pares de cromosomas se describen y nombran en función de su tamaño, del par 1 al 22 (de mayor a menor), más el par de cromosomas sexuales antes mencionado. El cromosoma 21 es el más pequeño, en realidad, por lo que debería ocupar el lugar 22, pero un error en la convención de Denver del año 1960, que asignó el síndrome de Down al par 21 ha perdurado hasta nuestros días, manteniéndose por razones prácticas esta nomenclatura.
El cromosoma 21 contiene aproximadamente el 1% de la información genética de un individuo en algo más de 400 genes, aunque hoy en día sólo se conoce con precisión la función de unos pocos.
En la imagen podemos observar un Cariotipo (conjunto de cromosomas de un individuo) mostrando una trisomía libre del par.
Trisomía libre
El síndrome de Down se produce por la aparición de un cromosoma más en el par 21 original (tres cromosomas: “trisomía” del par 21) en las células del organismo. La nomenclatura científica para ese exceso cromosómico es 47, XX,+21 o 47, XY,+21; según se trate de una mujer o de un varón, respectivamente. La mayor parte de las personas con este síndrome (95%), deben el exceso cromosómico a un error durante la primera división meiótica (aquella por la que los gametos, óvulos o espermatozoides, pierden la mitad de sus cromosomas) llamándose a esta variante, “trisomía libre” o regular. El error se debe en este caso a una disyunción incompleta del material genético de uno de los progenitores. (En la formación habitual de los gametos el par de cromosomas se separa, de modo que cada progenitor sólo transmite la información de uno de los cromosomas de cada par. Cuando no se produce la disyunción se transmiten ambos cromosomas).
No se conocen con exactitud las causas que originan la disyunción errónea. Como en otros procesos similares se han propuesto hipótesis multifactoriales (exposición ambiental, envejecimiento celular…) sin que se haya conseguido establecer ninguna relación directa entre ningún agente causante y la aparición de la trisomía. El único factor que presenta una asociación estadística estable con el síndrome es la edad materna, lo que parece apoyar las teorías que hacen hincapié en el deterioro del material genético con el paso del tiempo.
Translocación del brazo corto del cromosoma 21 en uno de los dos cromosomas del par 14.
Translocación del brazo corto del cromosoma 21 en uno de los dos cromosomas del par 14.
En aproximadamente un 15% de los casos el cromosoma extra es transmitido por el espermatozoide y en el 85% restante por el óvulo.
Translocación
Después de la trisomía libre, la causa más frecuente de aparición del exceso de material genético es la translocación. En esta variante el cromosoma 21 extra (o un fragmento del mismo) se encuentra “pegado” a otro cromosoma (frecuentemente a uno de los dos cromosomas del par 14), por lo cual el recuento genético arroja una cifra de 46 cromosomas en cada célula. En este caso no existe un problema con la disyunción cromosómica, pero uno de ellos porta un fragmento “extra” con los genes del cromosoma “translocado”. A efectos de información genética sigue tratándose de una trisomía 21 ya que se duplica la dotación genética de ese cromosoma.
La frecuencia de esta variante es aproximadamente de un 3% de todos los SD y su importancia estriba en la necesidad de hacer un estudio genético a los progenitores para comprobar si uno de ellos era portador sin saberlo de la translocación, o si ésta se produjo por primera vez en el embrión. (Existen portadores “sanos” de translocaciones, en los que se recuentan 45 cromosomas, estando uno de ellos translocado, o pegado, a otro).
Mosaicismo
La forma menos frecuente de trisomía 21 es la denominada “mosaico” (en torno al 2% de los casos). Esta mutación se produce tras la concepción, por lo que la trisomía no está presente en todas las células del individuo con SD, sino sólo en aquellas cuya estirpe procede de la primera célula mutada. El porcentaje de células afectadas puede abarcar desde unas pocas a casi todas, según el momento en que se haya producido la segregación anómala de los cromosomas homólogos.
Expresión del exceso de material genético (o sea lo que provoca este síndrome biológicamente)
La expresión bioquímica del síndrome consiste en el aumento de diferentes enzimas. Una de las más conocidas e importantes es la superóxidodimutasa (codificada por el gen SOD-1), que cataliza el paso del anión superóxido hacia peróxido de hidrógeno. En condiciones normales esto contribuye al sistema de defensa antioxidante del organismo, pero su exceso determina la acumulación de H2O2, lo que puede provocar peroxidación de lípidos y proteínas y dañar el ADN. Otros genes implicados en la aparición de trastornos asociados al SD son:
* COL6A1: Su expresión incrementada se relaciona con defectos cardiacos
* ETS2: Su expresión incrementada puede ser causa de alteraciones músculo esqueléticas
* CAF1A: La presencia incrementada de este gen puede interferir en la síntesis de ADN
* Cystathione Beta Synthase (CBS): Su exceso puede causar alteraciones metabólicas y de los procesos de reparación del ADN
* DYRK: En el exceso de proteínas codificadas por este gen parece estar el origen del retraso mental
* CRYA1: Su sobreexpresión puede originar cataratas (opacidad precoz del cristalino)
* GART: La expresión aumentada de este gen puede alterar los procesos de síntesis y reparación del ADN
* IFNAR : Es un gen relacionado con la síntesis de Interferón, por lo que su exceso puede provocar alteraciones en el sistema inmunitario.
Cuadro Clínico
El SD es la causa más frecuente de discapacidad psíquica congénita. Representa el 25% de todos los casos de retraso mental. Se trata de un síndrome genético más que de una enfermedad según el modelo clásico, y aunque sí se asocia con frecuencia a algunas patologías, la expresión fenotípica final es muy variada de unas personas a otras. Como rasgos comunes se pueden reseñar su fisiognomía peculiar, una hipotonía muscular generalizada, un grado variable de retraso mental y retardo en el crecimiento.
En cuanto al fenotipo han sido descritos más de 100 rasgos peculiares asociados al SD, pudiendo presentarse en un individuo un número muy variable de ellos. De hecho ninguno se considera constante o patognomónico aunque la evaluación conjunta de los que aparecen suele ser suficiente para el diagnóstico.
Algunos de los rasgos más importantes son un perfil facial y occipital planos, braquiocefalia (predominio del diámetro transversal de la cabeza), hendiduras palpebrales oblicuas, diastasis de rectos (laxitud de la musculatura abdominal), raíz nasal deprimida, pliegues epicánticos (pliegue de piel en el canto interno de los ojos), cuello corto y ancho con exceso de pliegue epidérmico nucal, microdoncia, paladar ojival, clinodactilia del quinto dedo de las manos (crecimiento recurvado hacia el dedo anular), pliegue palmar único, y separación entre el primer y segundo dedo del pie. Las patologías que se asocian con más frecuencia son las cardiopatías congénitas y enfermedades del tracto digestivo (celiaquía, atresia/estenosis esofágica o duodenal, colitis ulcerosa...). Los únicos rasgos presentes en todos los casos son la atonía muscular generalizada (falta de un tono muscular adecuado, lo que dificulta el aprendizaje motriz) y el retraso mental aunque en grados muy variables.
Presentan, además, un riesgo superior al de la población general, para el desarrollo de patologías como leucemia (leucemia mieloide aguda), diabetes, hipotiroidismo, miopía, o luxación atloaxoidea (inestabilidad de la articulación entre las dos primeras vértebras, atlas y axis, secundaria a la hipotonía muscular y a la laxitud ligamentosa). Todo esto determina una media de esperanza de vida entre los 50 y los 60 años, aunque este promedio se obtiene de una amplia horquilla interindividual (las malformaciones cardíacas graves o la leucemia, cuando aparecen, son causa de muerte prematura). El grado de discapacidad intelectual también es muy variable, aunque se admite como hallazgo constante un retraso mental ligero o moderado. No existe relación alguna entre los rasgos externos y el desarrollo intelectual de la persona con SD.
Epidemiologia
La incidencia global del síndrome de Down se aproxima a uno de cada 700 nacimientos (15/10.000), pero el riesgo varía con la edad de la madre. La incidencia en madres de 25 años es de 1 por cada 2000 nacidos vivos, mientras que en madres de 35 años es de 1 por cada 200 nacimientos y de 1 por cada 40 en las mujeres mayores de 40 años. Por este motivo se recomiendan técnicas de diagnóstico prenatal a todas las mujeres a partir de los 35 años. El ECEMC (Estudio Colaborativo Español de Malformaciones Congénitas) informaba en el año 2004 de una prevalencia neonatal de 7,11 cada 10.000 recién nacidos, con tendencia a disminuir de manera estadísticamente significativa. Esta tendencia, junto con el aumento relativo de casos en mujeres por debajo de 35 años, se atribuye al aumento de interrupciones voluntarias del embarazo tras el diagnóstico prenatal en mujeres por encima de esa edad. Parece existir una relación estadística (sin que se conozcan los mecanismos exactos) entre algunas enfermedades maternas como hepatitis, Mycoplasma hominis tipo 1, Herpes simple tipo II y diabetes y un aumento en la incidencia de aparición de SD; no obstante esa relación estadística no es tan intensa como en el caso de la edad materna. Algún autor también ha relacionado la baja frecuencia coital, así como el uso de anovulatorios y espermicidas con la aparición del síndrome.
Haber tenido un hijo con SD también aumenta la probabilidad de tener otro: el riesgo aumenta de modo general a 1 de cada 100 recién nacidos vivos. Los antecedentes familiares igualmente incrementan ese riesgo. Los varones con síndrome de Down se consideran estériles, pero las mujeres conservan con frecuencia su capacidad reproductiva. En su caso también se incrementa la probabilidad de engendrar hijos con SD.
DIAGNOSTICO
A partir de 1979 se dispone en los laboratorios de una prueba en sangre que permite establecer una sospecha diagnóstica para varios defectos congénitos (espina bífida y otros defectos del tubo neural). Esta prueba es la determinación de los valores de AFP (Alfa-fetoproteína), que se encuentran aumentados en los embriones que presentan estos trastornos del desarrollo. Varios años después se establece una relación estadística entre valores bajos de esta proteína y la aparición de trastornos cromosómicos, en especial del SD. En años posteriores se descubrieron algunas asociaciones similares con otras sustancias en sangre materna. Hoy día es común la determinación de AFP, estriol y hCG (Gonadotropina coriónica humana) para determinar el riesgo de aparición del SD. A esto se le llama “triple prueba”. Algunos laboratorios incluyen la determinación de inhibina (cuádruple prueba). Los valores de estas sustancias en sangre, así como datos acerca de la edad materna y los antecedentes personales y familiares permiten calcular un riesgo de aparición de SD, pero no suponen un diagnóstico de certeza. Determinadas mediciones que se realizan durante las ecografías (longitud del fémur, grosor del pliegue nucal, y otras) también aportan información para el cálculo de ese riesgo, pero tampoco permiten establecer el diagnóstico definitivo.
Para detectar la anormalidad cromosómica durante el periodo prenatal de forma inequívoca se emplean técnicas de conteo cromosómico, por lo que es necesario disponer de alguna célula fetal. El acceso al material celular embrionario puede suponer un cierto riesgo, tanto para la madre como para el feto, por lo que su indicación se circunscribe a aquellos embarazos en los que se haya detectado un riesgo de aparición de la trisomía superior al de la población general (triple prueba positiva, edad materna superior a 35 años o paterna superior a 50, antecedentes familiares o personales de SD, o progenitores portadores de una traslocación equilibrada u otras alteraciones cromosómicas).
La técnica más frecuentemente utilizada para la obtención de material genético fetal es la Amniocentesis. Esta técnica se empezó a generalizar en la década de los 60, y consiste en la punción ecoguiada de la cavidad amniótica por vía abdominal. Se consigue así una muestra de líquido amniótico, de donde es posible obtener células fetales para su estudio. Debe realizarse preferentemente entre las semanas 14 a 17 del embarazo. Es una técnica relativamente inocua y poco molesta pero comporta un riesgo del 1-2% de aborto, lesión fetal, o infección materna.
A mediados de los 80 se comenzó a usar otra técnica, denominada Biopsia de vellosidades coriónicas: se obtiene un fragmento de material placentario por vía vaginal o a través del abdomen, normalmente entre las semanas 8 y 11 del embarazo. Esta técnica se puede realizar antes de que exista la suficiente cantidad de líquido amniótico necesaria para que se pueda llevar a cabo la amniocentesis, y el estudio cromosómico es más rápido pues no se necesita el cultivo celular para obtener una muestra suficientemente grande. Presenta un riesgo para la madre y el feto similar al de la amniocentesis.
TRATAMIENTO
La mejoría en los tratamientos de las enfermedades asociadas al SD ha aumentado la esperanza de vida de estas personas, desde los 14 años hace unas décadas, hasta casi la normalidad (60 años, en países desarrollados) en la actualidad. A lo largo de los últimos 150 años se han postulado diferentes tratamientos empíricos (hormona tiroidea, hormona del crecimiento, ácido glutámico, dimetilsulfóxido, complejos vitamínicos y minerales, 5-hidroxitriptófano o piracetam) sin que ninguno haya demostrado en estudios longitudinales a doble ciego que su administración provoque ningún efecto positivo significativo en el desarrollo motor, social, intelectual o de expresión verbal de las personas con SD. No existe hasta la fecha ningún tratamiento farmacológico eficaz para el SD, aunque los estudios puestos en marcha con la secuenciación del genoma humano permiten augurar una posible vía de actuación (enzimática o genética), eso sí, en un futuro todavía algo lejano.
Los únicos tratamientos que han demostrado una influencia significativa en el desarrollo de los niños con SD son los programas de Atención Temprana, orientados a la estimulación precoz del sistema nervioso central durante los seis primeros años de vida. Especialmente durante los dos primeros años el SNC presenta un grado de plasticidad muy alto lo que resulta útil para potenciar mecanismos de aprendizaje y de comportamiento adaptativo. Los individuos con grandes dificultades para el aprendizaje a menudo han sido internados en instituciones, pero se ha comprobado que deben vivir en su domicilio, donde desarrollan de forma más completa todo su potencial. La adaptación curricular permite en muchos casos una integración normalizada en colegios habituales, aunque deben tenerse en cuenta sus necesidades educativas especiales. La edad mental que pueden alcanzar está todavía por descubrir, y depende directamente del ambiente educativo y social en el que se desarrollan. Cuando éste es demasiado protector, los chicos y chicas tienden (al igual que nos ocurriría a todos) a dejarse llevar, descubriendo escasamente sus potencialidades. Los contextos estimulantes ayudan a que se generen conductas de superación que impulsan el desarrollo de la inteligencia. Como consecuencia, es imposible determinar los trabajos y desempeños que pueden conseguir durante la vida adulta. Potenciar sus iniciativas y romper con los planteamientos estáticos que históricamente les han perseguido son compromisos sociales ineludibles que las sociedades actuales deben atender.
Atencion Temprana
Todos los niños precisan de estímulos para el correcto desarrollo de sus capacidades motrices, cognitivas, emocionales y adaptativas. Los niños con SD no son una excepción, aunque sus procesos de percepción y adquisición de conocimientos son algo diferentes a los del resto de la población: Las capacidades visuales de los niños con SD son, por ejemplo, superiores a las auditivas, y su capacidad comprensiva es superior a la de expresión, por lo que su lenguaje es escaso y aparece con cierto retraso, aunque compensan sus deficiencias verbales con aptitudes más desarrolladas en lenguaje no verbal, como el contacto visual, la sonrisa social o el empleo de señas para hacerse entender. La atonía muscular determina también diferencias en el desarrollo de la habilidad de caminar, o en la motricidad fina. Todos esos aspectos deben ser contemplados en programas específicos de atención temprana (durante los primeros seis años de vida) para estimular al máximo los mecanismos adaptativos y de aprendizaje más apropiados. Intentar enseñar a leer a un niño con SD utilizando métodos convencionales, por ejemplo, puede convertirse en una tarea muy difícil, si no se tiene en cuenta su superior capacidad visual. Hoy día existen métodos gráficos (a partir de tarjetas, o fichas, que asocian imagen y palabra) que están consiguiendo resultados muy superiores al clásico encadenado de letras en estos niños. Además el objetivo de estos programas no es tan sólo la adquisición de habilidades, sino que estas se alcancen mucho antes, permitiendo continuar con programas educativos que integren al máximo a la persona con SD en entornos normalizados
[b○3Pronosticos y expectativas de futuro[/b]
Se desconocen todavía los mecanismos que provocan el retraso mental en las personas con SD, aunque la secuenciación del genoma humano y diversos estudios llevados a cabo en sujetos con translocaciones parciales están empezando a servir para descubrir los genes responsables del cuadro. Estos mapas fenotípicos también se han comparado con algunos casos de monosomía 21 (cuadro de ausencia de uno de los dos cromosomas del par 21, la situación contraria al SD) obteniéndose así mapas de rasgos asociados al exceso o defecto de dosis cromosómica. [36] En las próximas décadas todo este conocimiento sobre el funcionamiento y expresión de los genes permitirá, con seguridad, establecer nuevas estrategias terapéuticas capaces de revertir los trastornos cognitivos asociados al síndrome de Down, y muchos de sus problemas asociados.
En 1981 se diseñó el primer Programa de Salud específico para personas con SD, pero el más ampliamente aceptado y difundido en la comunidad científica es el diseñado por el Down Syndrome Medical Interest group (DSMIG) . En estos programas de salud se contemplan las actuaciones preventivas mínimas para un adecuado diagnóstico precoz y seguimiento de las enfermedades o complicaciones que se pueden presentar, mejorando significativamente el pronóstico de estas personas. Por otra parte los programas, cada vez más extendidos, de estimulación precoz, y el cambio progresivo de mentalidad que la sociedad está experimentando con respecto a la discapacidad intelectual son los principales motivos de la gran transformación que se está viviendo en torno a las personas con SD. Hace apenas unas décadas estas personas eran apartadas de la sociedad en instituciones, o escondidas por sus progenitores, en base a un falso complejo de culpa. A pesar del enorme esfuerzo que aún queda pendiente hoy podemos comprobar cómo un entorno basado en la aceptación, en la adaptación de los métodos de aprendizaje y en la virtud de la diversidad está dotando a las personas con SD de la autonomía suficiente como para trabajar, vivir en pareja o desarrollar habilidades artísticas impensables hace muy poco tiempo
Ufff ahora si el video de Sigur Ros
SIGUR ROS - Svefn-g-englar
link: http://www.videos-star.com/watch.php?video=zQ5Grncdjlc
Frases al principio del video
"Sin música la vida seria una equivocación" (Friedrich Nietzche)
"La música fue inventada para confirmar la soledad humana" (Lawrence Dureeus)
"La música es la forma rápida y abreviada de las emociones" [Leo Toisley)
"La música expresa aquella que no puede ser dicho en lo que es imposible de callar" (Victor Hugo)
"La música es una ley moral, le da alma al universo, alas a la mente, vuelo a la imaginación, encanto y tranquilidad a la vida y a todo" (Platón)
"Un pintor pinta imágenes en lienzos. Pero los músicos pintan su música en el silencio" (Nombre Ilegible)
"Si miras lo suficientemente profundo, verás musicalmente. El corazón de la Naturaleza siendo música en todos lados" (Thomas Cartile)
"La música nombra lo innombrable y comunica lo imposible de conocer" (Leonard Bernstein)
"...En el final veo a la música como la gracia de salvación para toda la humanidad" (Henry Millers)
"La música es bien nombrada como el habla de los ángeles, de hecho nada entre los (esta palabra se refiere a un acto no verbal en el que se transmite un mensaje, como un lenguaje no hablado) permitidos al hombre se siente tan divino. Nos lleva cerca del infinito" (Thomas Carlyle)
Ahora si la fuente... es wikipedia pero citare las fuentes que usaron los users en wikipedia, he revisado y son confiables entre a los links y revise si estan muertos y arregle algunos que estaban mal, todas las fuentes 100% confiables y comprobadas por moi
DADO LO EXTENSO ME HE TOMADO LA LIBERTAD DE RESALTAR ALGUNOS ASPECTOS QUE DEBERIAN SER LEIDOS.
* Siegfried M. Pueschel (2002) Síndrome de Down: Hacia un futuro mejor, Ed. Masson ISBN 1-55766-452-8.
* Down, J.H.L. (1886). Observations on an ethnic classification of idiots. London Hospital. Clinical Lectures and Reports, 3: 259-262.
* Josep M. Corretger et al (2005). Síndrome de Down: Aspectos médicos actuales. Ed. Masson, para la Fundación Catalana del Síndrome de Down. ISBN 84-458-1504-0.
* Azucena Martínez Acebal, Joaquín Fernández Toral (1999). Síndrome de Down: Aspectos sociológicos, Médicos y Legales. ISBN 84-86889-65-0.
* Pilar Arranz Martínez (2002). Niños y jóvenes con Síndrome de Down. Egido Editorial. ISBN 84-95879-09-3.
* Candel, I. Programa de Atención temprana. Intervención en niños con síndrome de Down y otros problemas del desarrollo. Ed. CEPE, Madrid, 1999.
http://www.ctdownsyndrome.org/index.php?option=com_content&task=view&id=73&Itemid=118

